Quick start
BRIE2 estimates the splicing proportion for two-component events across many cells. It is designed for analysing two molecular levels of splicing processes:
Alternative splicing isoforms, e.g., exon included or excluded. BRIE2 will identify differential alternative splicing (DAS) events. See demo1 for detecting DAS between disease and control.
Spliced vs unspliced RNAs for RNA velocity analysis. BRIE2 will identify differential momentum genes (DMG). See demo2 for detecting DMG between cell types.
For getting started quickly, there are two steps to go: counting and
quantifying.
The brie-quant (for quantifying and testing) is developed in a unified
manner. Here we provide a roadmap of using BRIE2 for both molecular levels
in different purposes (quantification or feature detection).
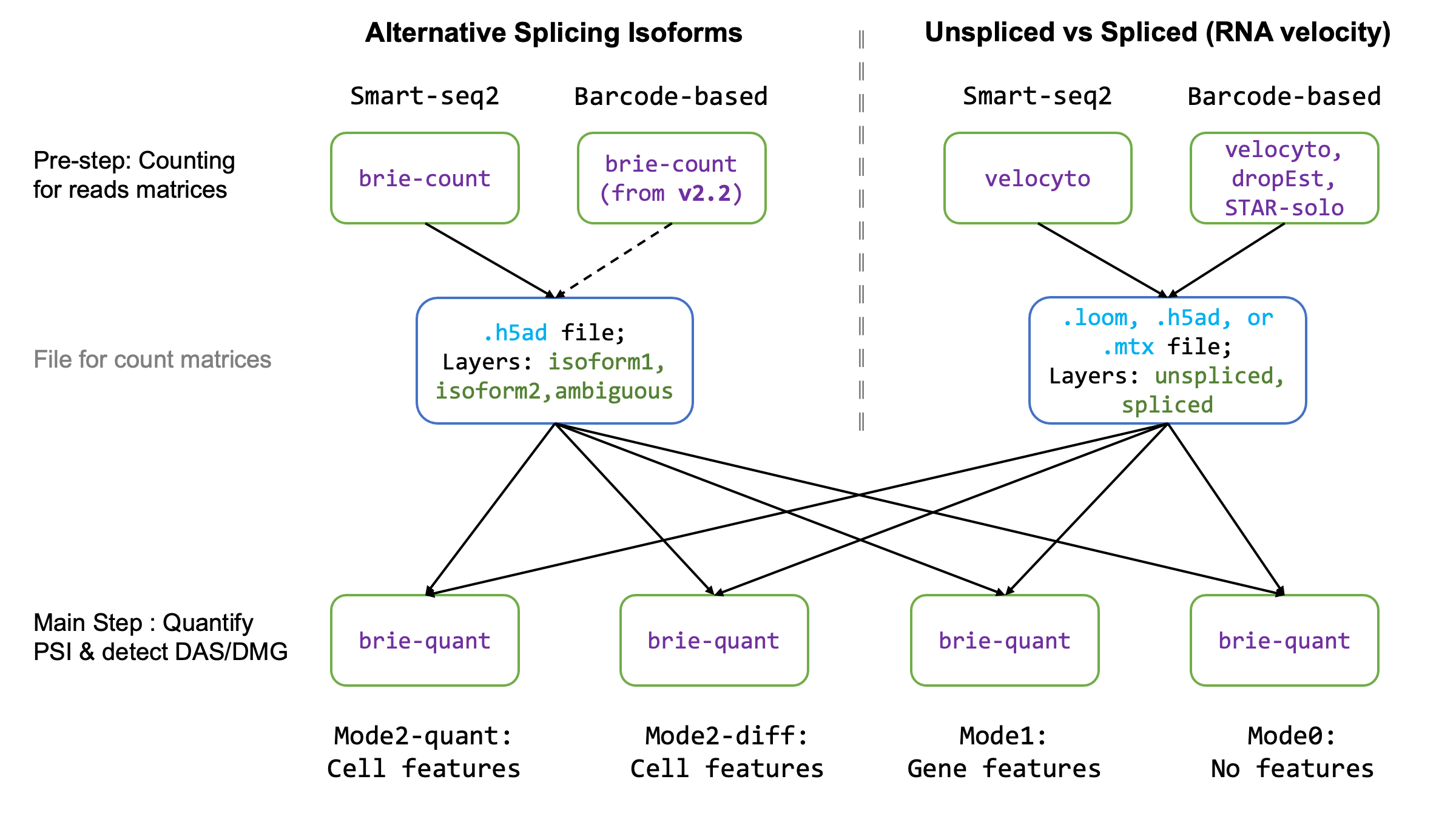
Pre-step: read counting
First, you need to count the isoform-specific reads in each splicing event in each cell. Similarly for read/UMI counts for spliced and unspliced RNAs. BRIE2 only provides a utility function for read counts in alternative splicing. For counting spliced/unspliced reads, we provide some tips in the unspliced RNA counting section.
For alternative splicing, e.g., exon-skipping event, you can download the splicing annotations generated by us or make your own, e.g., with briekit.
Then you can use the brie-count to fetch the read count tensor, which will
be stored in hdf5 format as AnnData. See more details on brie-count
CLI, and you can use this example command line.
# for smart-seq
brie-count -a AS_events/SE.gold.gtf -S sam_and_cellID.tsv -o out_dir -p 15
# for droplet, e.g. 10x Genomics
brie-count -a AS_events/SE.gold.gtf -s possorted.bam -b barcodes.tsv.gz -o out_dir -p 15
Besides the SE event, other types of alternative splicing, e.g., intron retaining is also applicable with BRIE. Some pre-processing utilities will be available soon.
Main Step: quantify and detect
brie-quant is used for this step. If GPU is available, we highly
recommend using GPU for ~10x speedup comparing CPU server. Thanks to good
support from Tensorflow, the environment setting is straightforward, see our
guide on GPU Usage.
This step supports various settings for either quantification of splicing or detection of splicing phenotypes. You can directly run step 1.2 if you only want to perform phenotype detection.
PSI quantification
You can quantify the isoform with cell or gene features or both or none. Usually,
we recommend using aggregated imputation even if you don’t have any feature,
namely mode2-quant in brie-quant CLI as follows (please add
--interceptMode gene for aggregating cells as prior for each gene),
brie-quant -i out_dir/brie_count.h5ad -o out_dir/brie_quant_aggr.h5ad --interceptMode gene
Splicing Phenotype detection
If you have cell-level features, e.g., disease condition or cell type or
continuous variable, you can use it in cell features to detect variable splicing
events or differential momentum genes as phenotypes for further analysis. This
is mode2-diff in brie-quant CLI, so requires -c and
--LRTindex.
brie-quant -i out_dir/brie_count.h5ad -o out_dir/brie_quant_cell.h5ad \
-c $DATA_DIR/cell_info.tsv --interceptMode gene --LRTindex=All
Example
Please see the example in brie-quant CLI mode 3, and MS data.
Downstream Step: analysis
The BRIE output AnnData files are compatible with Scanpy, hence you can easily use it for dimension reduction, clustering, and other visualization. A few examples for both alternative splicing and RNA velocity are also available in this documentation (see the navigation bar on the left).
Others for unspliced RNA counting
BRIE2 doesn’t provide a utility function for counting the spliced and unspliced RNAs, but thanks to the community efforts, there are a few tools already available for this purpose:
velocyto.py: the earliest software for this purpose. Generally not computationally efficient, possible due to written in Python. For unknown reasons, the proportion of unspliced RNA is unrealistically high for 5’ scRNA-seq data based on 10x Genomics.
dropEst: as implemented in C/C++, it is much more efficient. It also returns more reasonable proportions of unspliced RNAs for 5’ 10x Genomics data
STAR-solo: new extension for the popular STAR. Benefits: efficient and one step for reads alignment and counting of unspliced RNA (Recommended option)
The first two options take inputs as aligned bam file(s), and STAR-solo itself is a widely used aligner and provides the count matrices directly. All these options align reads to genome and define reads as unspliced and spliced by the gene annotations in GTF/GFF3 format.
Alternatively, there are other options by aligning reads to annotated transcriptomes directly e.g., kallisto bustools. However, the agreement of the above counting tools is still not perfect according to a recent benchmarking paper (Soneson et al, Plos Comp Bio, 2021)